| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
You are here Biopharmaceutical/ Genomic Glossary homepage > Technologies > Sequencing
Sequencing Glossary & taxonomy The "race" to sequence the Human Genome was not a 100 yard dash, but a marathon. Although the Human Genome Project finished well ahead of schedule, and a number of genes have been identified, we have just begun to get a glimpse of what specific genes do and how we might be able to better use this knowledge for therapeutic interventions. Teasing apart the interactions of genes and proteins, delineating changes throughout the cell cycle, and correlating changes with health and disease will take even more time. But with complete sequences, and the cross- species comparisons we can expect new insights and speeding up over time. Sequencing DNA is only a first step towards finding what functions are connected with specific sequences. Sequencing proteins (and determining the structures – and functions of proteins) is ongoing.
Related glossaries include
Biomarkers Molecular
Diagnostics Molecular
Medicine $1,00 genome : Molecular Diagnostics
Clinical genome sequencing explores
the recent surge in clinical genome sequencing, from the point of view of
the sequencing providers, the medical organizations delivering these
services, and the start-ups offering a variety of interpretation services,
platforms, and business models. Aspects include: Progress in clinical
genome sequencing, Organizations leading the way in generating clinical
data and its interpretation, Determining the causality of documented
variants in genetic disease, Clinical genome sequencing in oncology,
Academic and commercial clinical genomics providers, next-gen sequencing
landscape, Companies providing genome interpretation software, Initiatives
in setting sequencing standards, custom survey results on clinical genome
sequencing.
Insight Pharma Reports Advances in
Clinical Genome Sequencing and Diagnostics 2013
de novo sequencing: Determination of sequences (of genes or amino acids) whose sequence is not yet known. Can be done with LC/MS/MS or nanoelectrospray MS/MS. From the Latin "de novo" from the beginning. See also Mass spectrometry
deep
sequencing: Techniques
of nucleotide sequence analysis that increase the range, complexity,
sensitivity, and accuracy of results by greatly increasing the scale of
operations and thus the number of nucleotides, and the number of copies of each
nucleotide sequenced. The sequencing may be done by analysis of the synthesis or
ligation products, hybridization to preexisting sequences, etc. MeSH 2011
Deep Sequencing and Single Cell Analysis for Antibody Discovery
Technologies and Best Practices for Applying Repertoire Analysis in the
Discovery of Therapeutic Proteins
JANUARY 23-24, 2020 San Diego CA
The rapid adoption of deep sequencing and single B cell analysis has given
discovery scientists an extraordinary view into human and animal immune
repertoires that is now informing all aspects of biopharmaceutical R&D.
This dynamic field is bringing together the disciplines of immunology,
structural and computational biology, informatics and microfluidics to
offer previously unimaginable perspectives that will drive discovery of
the next generation of biologic drugs
genotype:
The genetic constitution of an organism as revealed
by genetic or molecular analysis, i.e. the complete set of genes, both
dominant and recessive, possessed by a particular cell or organism. IUPAC
Biotech
The observed alleles at a genetic locus for an individual.
NHLBI The genetic constitution of the individual; the
characterization of the genes. MeSH 1968
genotyping:
The determination of relevant nucleotide- base sequences
in each of the two parental chromosomes. May
refer to identifying one or more, up to the entire gene sequence of an
organism. Compare phenotype. Used for diagnosis,
drug efficacy, and toxicity. Utilizes genomic DNA
that, after digestion, reacts with a SNP array to
obtain an individual SNP pattern. These variations
can for instance provide information about the
diagnosis of a certain disease, or the
effectiveness or side effect of a certain drug.
Genotyping implies (though I haven't found this in print) determining known
variants, as opposed to discovery of new ones. Related terms SNPS & other
genetic
variations; Broader term sequencing;
Narrower terms: haplotyping, genome wide association studies
GWAS
Genome Wide Association Sequencing: Genomic
informatics
haplogroups:
The term 'haplogroup'
refers to the SNP/unique-event
polymorphism (UEP) mutations that represent the clade to
which a collection of particular human haplotypes belong. (Clade here refers to
a set of haplotypes sharing a common ancestor.)[7] A haplogroup is
a group of similar haplotypes that share a common ancestor with a single-nucleotide
polymorphism mutation.[8][9] Mitochondrial
DNA passes along a maternal
lineage that can date back thousands of years.[8]
Wikipedia accessed 2018 Nov 8
https://en.wikipedia.org/wiki/Haplotype
(haploid from
the Greek: ἁπλούς, haploûs,
"onefold, simple" and English: group)
is a group of similar haplotypes that share a common ancestor with a single-nucleotide
polymorphism mutation.[3][4] More
specifically, a haplogroup is a combination of alleles at
different chromosomes regions that are closely linked and that tend to be
inherited together. As a haplogroup consists of similar haplotypes, it is
usually possible to predict a haplogroup from haplotypes. Haplogroups
pertain to a single line
of descent. As such, membership of a
haplogroup, by any individual, relies on a relatively small proportion of
the genetic material possessed by that individual.
Wikipedia accessed 2018 Nov 8 http://en.wikipedia.org/wiki/Haplogroup
haplotype:
The genetic constitution of individuals with respect to one member of a pair
of allelic genes, or sets of genes that are closely linked and tend to be
inherited together such as those of the MAJOR HISTOCOMPATIBILITY COMPLEX. MeSH,
1987
a group of alleles in
an organism that
are inherited together from a single parent.[1][2] However,
there are other uses of this term. First, it is used to mean a collection
of specific alleles (that is, specific DNA sequences)
in a cluster of tightly linked genes on a chromosome that
are likely to be inherited together—that is, they are likely to be conserved
as a sequence that survives the
descent of many generations of reproduction.[3][4] A
second use is to mean a set of linked single-nucleotide
polymorphism (SNP) alleles that tend
to always occur together (i.e., that are associated
statistically). It is thought that
identifying these statistical associations and few alleles of a specific
haplotype sequence can facilitate identifying all other such polymorphic
sites that are nearby on the chromosome. Such information is critical for
investigating the genetics of common diseases;
which in fact have been investigated in humans by the International
HapMap Project.[5][6] Thirdly,
many human genetic testing companies use the term in a third way: to refer
to an individual collection of specific mutations within a given genetic
segment; (see short
tandem repeat mutation). Wikipedia
accessed 2018 Nov 8
https://en.wikipedia.org/wiki/Haplotype
A haplotype is the set
of SNP alleles along a region of a chromosome. Theoretically there could be many
haplotypes in a chromosome region, but recent studies are typically finding only
a few common haplotypes. Developing a Haplotype map of the human genome, 2001 http://www.genome.gov/10001665
A particular pattern of sequential SNPs
found on a single chromosome. These SNPs tend to be inherited together over time
and can serve as disease-gene markers. The examination of single chromosome sets
(haploid sets), as opposed to the usual chromosome pairings (diploid sets), is
important because mutations in one copy of a chromosome pair can be masked by
normal sequences present on the other copy.
From “haploid genotype.” The
key idea is that alleles often travel together. Related terms: haplotyping, haplotyping technologies
Cell biology diploid, haploid, ploidy; Maps
& mapping:
haplotype map HapMap; Narrower term: SNPs
& genetic
variations haploinsufficiency, haplotype block, SNP haplotype
Somatic cells, as opposed to germ
cells, have two copies of each chromosome. A given single- base position may be homozygous
for the wild- type base (each chromosome has the normal allele),
homozygous for a SNP base (each chromosome has the altered allele), or
heterozygous for two different bases (one chromosome has the normal allele
and the other has the abnormal allele). Haplotyping involves grouping
subjects by haplotypes, or particular patterns of sequential SNPs, found
on a single chromosome. These SNPs tend to be inherited together over time and
can serve as disease- gene markers. The examination of single chromosome sets (haploid
sets), as opposed to the usual chromosome pairings (diploid sets), is
important because mutations in one copy of a chromosome pair can be masked by
normal sequences present on the other copy. Genes tend to travel
in packs. This is good news for pharmacogenomics.
Broader
terms genotyping, sequencing
haplotyping technologies: Include
microarrays, mass
spectrometry, sequencing
immunosequencing:
a
platform technology that allows the enumeration, specification and
quantification of each and every B-and/or T-cell in any biologic sample of
interest. It is based on bias-controlled multiplex PCR and high throughput
sequencing and is highly accurate, standardized, and sensitive.
Immune
monitoring technology primer: immunosequencing,
Ilan Kirsch
J Immunother Cancer. 2015;
3: 29. Published online 2015 Jun 25. doi: 10.1186/s40425-015-0076-y
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4480450/
Maxam-Gilbert sequencing & Sanger sequencing:
a method of DNA
sequencing developed by Allan
Maxam and Walter
Gilbert in 1976–1977. This method is based on nucleobase-specific
partial chemical modification of DNA and subsequent cleavage of
the DNA backbone at sites adjacent to the modified nucleotides.[1]
… Maxam–Gilbert sequencing
was the first widely adopted method for DNA sequencing, and, along with
the Sanger
dideoxy method, represents the first generation of DNA
sequencing methods. Maxam–Gilbert sequencing is no longer in widespread
use, having been supplanted by next-generation sequencing methods.
Wikipedia accessed 2018 March 16
https://en.wikipedia.org/wiki/Maxam%E2%80%93Gilbert_sequencing
microsequencing:
Sequencing of proteins or peptides in very small
amounts (sub microgram), sometimes for use as probes.
minisequencing: A solid- phase method for the detection of any
known point mutation or allelic variation of DNA. In the method amplified,
biotinylated DNA sequences containing the mutation site are immobilized
onto streptavidin coated microplate and primer extension reactions are
carried out using labeled nucleotides. Incorporation of the labeled nucleotide
is dependent on the genotype and is analyzed using ELISA technique. Assay
method allows automation. Photometry applications, Labsystems Oy, Finland, no
longer on website Single base sequencing.
multilocus sequence
typing: Direct nucleotide sequencing of gene fragments from multiple
housekeeping genes for the purpose of phylogenetic analysis, organism
identification, and typing of species, strain, serovar, or other distinguishable
phylogenetic level. MeSH 2011
nanopore
sequencing: a
third generation[1] approach
used in the sequencing of biopolymers-
specifically, polynucleotides in
the form of DNA or RNA.
Wikipedia accessed 2018 Sept 4
next generation
sequencing: High-throughput (formerly
"next-generation") sequencing applies to genome sequencing, genome
resequencing, transcriptome profiling
(RNA-Seq),
DNA-protein interactions (ChIP-sequencing),
and epigenome characterization.[55]. Resequencing
is necessary, because the genome of a single individual of a species will not
indicate all of the genome variations among other individuals of the same
species. The high demand for
low-cost sequencing has driven the development of high-throughput sequencing
technologies that parallelize the sequencing process, producing
thousands or millions of sequences concurrently.[56][57][58] High-throughput
sequencing technologies are intended to lower the cost of DNA sequencing beyond
what is possible with standard dye-terminator methods.[59] In
ultra-high-throughput sequencing as many as 500,000 sequencing-by-synthesis
operations may be run in parallel.[60][61][62]
Wikipedia accessed 2018 March 16
https://en.wikipedia.org/wiki/DNA_sequencing#Next-generation_methods
optical
mapping: Stretching
DNA molecules in nanochannels allows structural and copy-number variations to be
visualized like beads on a string. Channeling
DNA for optical mapping
Yael
Michael & Yuval
Ebenstein Nature Biotechnology 30,
762–763 (2012) doi:10.1038/nbt.2324 published online August
Next
generation sequencing (NGS) is revolutionizing all fields of biological research
but it fails to extract the full range of information associated with genetic
material. Optical mapping of DNA grants access to genetic and epigenetic
information on individual DNA molecules up to ∼1 Mbp
in length. Beyond
sequencing: optical mapping of DNA in the age of nanotechnology and nanoscopy Michal
Levy-Sakin, Yuval
Ebenstein Current
Opinion in Biotechnology Volume
24, Issue 4, August 2013, Pages 690–698 http://www.sciencedirect.com/science/article/pii/S0958166913000128
pathogen sequencing:
In the future, more pathogens will have their
genomes completely sequenced to determine not only how the pathogen causes
disease, but what, if any, treatments will be most effective. The DNA sequences
of viruses like HIV, human papilloma virus (HPV), and hepatitis C (HCV) are
already being characterized and therapies prescribed based on this genetic
information. To perform these types of diagnoses, DNA sequencing will have to
become faster, more cost effective, simpler to perform, and more accessible to
clinical laboratories.
PCR
and NGS-Based Molecular Diagnostics
March 14-15, 2019 San Francisco, CA
Program |
Advances
Techniques and Tools for Precision Medicine
Advances in molecular
diagnostics technologies have sparked innovation, expanded research
capabilities, and enhanced clinical diagnostics. Cambridge Healthtech
Institute’s 6th Annual PCR and NGS-Based Diagnostics symposium puts an
emphasis on the NGS and PCR technologies that drive precision medicine and
showcases how they are being used to alter clinical outcomes. This event
will provide a comprehensive look at integrating molecular diagnostics
solutions for biomarker discovery and development, point-of-care,
companion diagnostics, and infectious disease.
published
working drafts - human genome:
International Human Genome Sequencing Consortium special issue: Nature
409 (6822) 15 Feb 2001 Human Genome [Celera Genomics sequence] special
issue: Science 291
(5507) Feb. 16, 2001 http://www.sciencemag.org/content/vol291/issue5507/index.shtml
resequencing:
Eric Lander, director of the Whitehead Institute's Center for Genome Research, and professor of biology at
MIT notes " The human genome will need to be sequenced only once, but it will be
resequenced thousands of times, in order, for example to unravel the polygenic
factors underlying human susceptibilities and predispositions … Re-sequencing
will also provide the ultimate tool for genotyping studies" E. Lander
"The New Genomics" Science 274: 536, 25 Oct. 1996
Previously sequenced site is resequenced for SNP
discovery or other purposes. DNA resequencing involves sequencing a DNA
region where a reference sequence for the region is already available. These
studies provide important insight into the function of genes and the evolution
of genes and populations. Applications abound including: comparative genomics,
high-throughput SNP detection, identifying mutant genes in disease pathways,
profiling transcriptomes for organisms where little information is available,
researching lowly expressed genes, to identifying newly emerging or genetically
engineered bacterial and viral strains.
RNA-Seq (RNA sequencing): also called whole
transcriptome shotgun sequencing[2] (WTSS),
uses next-generation
sequencing(NGS) to reveal the presence and quantity of RNA in
a biological sample at a given moment.[3][4]
RNA-Seq is used to analyze the continuously changing cellular transcriptome.
Wikipedia accessed 2018 Oct 21
https://en.wikipedia.org/wiki/RNA-Seq
scanning, scoring: SNPs
& other genetic variations sequence
coverage: refers to the general concept of aiming for high number
of unique reads of each region of a sequence.[3]
Wikipedia accessed 2018 Aug 28
sequence inversion:
The
deletion and reinsertion of a segment of a nucleic acid sequence in the same
place, but flipped in an opposite orientation. MeSH 2010
sequencing:
Proteins, nucleic acids
-- Analytical procedures for
the determination of the order of amino acids in a polypeptide chain or
of nucleotides in a DNA or RNA molecule. IUPAC Compendium
Largely automated now. Full DNA sequencing is the "gold standard"
for genotyping. Narrower terms; next generation
sequencing, shotgun sequence, de novo sequencing, microsequencing,
minisequencing, multiplex sequencing, Sanger sequencing, sequencing by
synthesis. Related terms: genotyping, GWAS Genome Wide Association
Sequencing, haplotyping, sequencing data analysis &
storage, sequencing data management
sequencing by
synthesis:
Promising
new sequencing technologies, based on sequencing by synthesis (SBS), are
starting to deliver large amounts of DNA sequence at very low cost.
Polymorphism detection is a key application. Quality
scores and SNP detection in sequencing-by-synthesis systems., Brockman W,
Alvarez P, Young S, Garber M, Giannoukos G, Lee WL, Russ C, Lander ES, Nusbaum
C, Jaffe DB, Genome Research 2008 Jan 22
The “sequencing-by-synthesis” technology now used by Illumina was
originally developed by Shankar Balasubramanian and David Klenerman at the
University of Cambridge. They founded the company Solexa in 1998 to
commercialize their sequencing method. Illumina went on to purchase Solexa
in 2007 https://bitesizebio.com/13546/sequencing-by-synthesis-explaining-the-illumina-sequencing-technology/
sequencing - cost of:
Cheap and easy genome sequencing has been both a
blessing and a curse. We are able to find an incredible wealth of variation, but
for the most part we have no easy way to tell whether a difference might
contribute to a disease or not. The poster child for this problem is autism.
Lots of genome wide association studies (GWAS) have been done and lots of rare
variants in lots of different genes have been found – unfortunately, way too
many to pick out the ones that really matter. Luckily our friend yeast can
help. Yeast winnows down GWAS hits in autism, SGD Database 2013 https://www.yeastgenome.org/blog/yeast-winnows-down-gwas-hits-in-autism
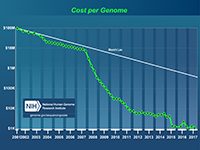
Related term: Molecular Diagnostics $1,000
genome
sequencing - high- throughput:
Uses robotics, automated DNA-
sequencing machines and computers.
shotgun sequencing:
Sequencing method which involves randomly sequencing
tiny cloned pieces of the genome, with no foreknowledge of where on a chromosome
the piece originally came from. This can be contrasted with "directed"
[sequencing] strategies, in which pieces of DNA from adjacent stretches of a chromosome
are sequenced. Directed strategies eliminate the need for complex reassembly
techniques. Because there are advantages to both strategies, researchers
expect to use both random (or shotgun) and directed strategies in combination
to sequence the human genome. DOE
third-generation
sequencing (TGS): Sequencing single DNA molecules without the need to
halt between read steps (whether enzymatic or otherwise).
A window into
third-generation sequencing, Glossary
Eric E. Schadt*, Steve Turner Andrew Kasarskis,
Human Molecular Genetics
19, IssueR2
Pp. R227-R240.
http://hmg.oxfordjournals.org/content/19/R2/R227
transcriptome sequencing:
Deep sequencing of transcriptomes,
also known as RNA-Seq,
provides both the sequence and frequency of RNA molecules that are present
at any particular time in a specific cell type, tissue or organ.[8] Counting
the number of mRNAs that are encoded by individual genes provides an
indicator of protein-coding potential, a major contributor to phenotype.[9] Improving
methods for RNA sequencing is an active area of research both in terms of
experimental and computational methods.[10]
Wikipedia accessed 2018 Aug 28
https://en.wikipedia.org/wiki/Coverage_(genetics)#Transcriptome_sequencing
viral genotyping:
Genomic
data is enabling researchers to predict a patient's response to therapy based on
the viral genotype for viral infections. HIV genotyping is an early example of
how treatment decisions are made based on the genotype of the virus.
Whole
Exome Sequencing Techniques to
determine the complete complement of sequences of all EXONS of an organism
or individual. MeSH 2018
Whole Genome
Sequencing: Techniques to
determine the entire sequence of the GENOME of an organism or individual.
Year introduced: 2018 MeSH
whole genome shotgun sequencing:
Whole Genome Shotgun (WGS) sequencing projects are
incomplete genomes or incomplete chromosomes that are being sequenced by a whole
genome shotgun strategy. WGS projects may be annotated, but annotation is not
required. The pieces of a WGS project are the contigs (overlapping reads), and
they do not include any gaps. NCBI Whole Genome Shotgun Submissions http://www.ncbi.nlm.nih.gov/genbank/wgs.html
Broader term shotgun sequencing method.
Related
term: GWAS Genome Wide Association Sequencing
Wikipedia http://en.wikipedia.org/wiki/Shotgun_sequencing#Whole_genome_shotgun_sequencing
Technologies Conferences
http://www.healthtech.com/conferences/upcoming.aspx?s=TCH IUPAC definitions are reprinted with the permission of the International
Union of Pure and Applied Chemistry.
|
Contact
| Privacy Statement |
Alphabetical
Glossary List | Tips & glossary
FAQs | Site Map